Pr. Sylvie DI FILIPPO
Centre Cardio-Thoracique de Monaco.
Cardiologia congenita pediatrica e adulti, cardiologia fetale – Professore delle Università.
I difetti cardiaci congeniti rappresentano le anomalie congenite d’organo più comuni. La loro incidenza nella popolazione in generale è di 14 feti ogni 1000 e 8 nati vivi ogni 1000.
Le cardiopatie congenite comprendono malformazioni multiple, di complessità anatomo-funzionale variabile e prognosi eterogenea.
Queste malformazioni vengono classificate in base alla loro gravità e complessità in:
- Semplici, moderate e severe
- Riparabili e non riparabili
- Biventricolari e univentricolari
- Cianogene e non cianogene
Cardiopatie minori
|
Cardiopatie moderate
|
Cardiopatie maggiori
|
|
|
|
Classificazione anatomo-fisiopatologica delle cardiopatie congenite
- Shunt sinistro-destro
- Shunt destro-sinistro e ostacoli destri
- Ostacoli lasciati
- Anomalie coronariche
- Anomalie del ritmo cardiaco
- Malattie cardiache complesse
Shunt: comunicazione anormale naturale o creata tra camere o vasi cardiaci.
Cianosi: colorazione bluastra della pelle e delle mucose dovuta a un calo di ossigeno nel sangue (o ipossia).
Screening prenatale
- I difetti cardiaci congeniti sono ben tollerati durante la vita intrauterina e non impediscono il normale sviluppo del feto.
- L’adattamento alla vita extrauterina, che comporta un calo della pressione polmonare e la chiusura del dotto arterioso e del forame ovale provoca lo scompenso delle malformazioni cardiache la cui vitalità dipende dalla permeabilità di questi shunt.
- La diagnosi prenatale della cardiopatia mediante ecocardiografia e Doppler consente di anticipare e prevenire il suo scompenso, guidando l’immediata condotta terapeutica postnatale. Il parto viene programmato e pianificato dall’équipe gineco-ostetrica e neonatologica.
- Nella maggior parte dei casi, l’induzione prematura non è necessaria. Solo i casi di insufficienza cardiaca fetale possono giustificarlo (conseguenza di un ritmo cardiaco anomalo, sia troppo lento = bradicardia fetale, sia troppo veloce = tachicardia fetale incontrollata).
- Lo screening prenatale consente la diagnosi di cardiopatie con una performance che varia dal 40% al 90% a seconda del tipo di cardiopatia
- Permette di stabilire una diagnosi, monitorare i progressi prenatali, pianificare l’assistenza postnatale e stabilire la prognosi
- Il parto così programmato aiuta a prevenire lo scompenso delle malattie cardiache dopo la nascita
- La somministrazione della Prostaglandina E1 è un trattamento che mantiene aperto il dotto arterioso alla nascita, mantenendo la circolazione e/o l’ossigenazione nei neonati con malattie cardiache a rischio di scompenso acuto.
- Le malattie cardiache a rischio di scompenso neonatale che comporta un rischio letale includono:
• Ostacoli diritti che limitano il flusso polmonare
• Ostacoli sinistri che limitano il flusso aortico
• Anomalie di mescolamento che dipendono dalla permeabilità del forame ovale.
Diagnosi e sintomi clinici
Neonato
- Cianosi, insufficienza cardiaca
- Cardiopatia shunt-dipendente
Sintomi
-
- Difficoltà di alimentazione
- Scarsa crescita o aumento delle infezioni broncopolmonari
- Dispnea da sforzo
- Malessere, sincope
- Dolore toracico
Asintomatico
- Respiro
- Riscontro accidentale di anomalie dell’ECG
- Anomalie all’esame clinico: polso femorale, ipertensione
Effettuare un’ecografia cardiaca e un Doppler: l’ecografia cardiaca è un esame semplice e non invasivo che può essere utilizzato per diagnosticare difetti cardiaci a qualsiasi età.
Neonatologia: screening per le cardiopatie congenite
Prenatale: la cardiopatia rilevata prima della nascita deve essere confermata dopo la nascita mediante ecografia cardiaca nel neonato.
Postnatale: non tutte le malattie cardiache vengono rilevate prima della nascita e l’esame di tutti i neonati nel reparto maternità resta molto importante per cercare un’eventuale anomalia cardiaca non precedentemente diagnosticata cosi da riconoscerla prima del ritorno a casa.
L’esame clinico deve ricercare:
- Palpazione dei polsi femorali
- La presenza di un soffio all’ausculazione
- Il comportamento alimentare del neonato
- Saturazione di ossigeno o satO2: lo screening sistematico per le cardiopatie neonatali viene attuato nei reparti maternità con la misurazione della saturazione di ossigeno negli arti superiori e inferiori, il cui tasso normale è > 97% con una differenza inferiore al 3% tra gli arti superiori e arti inferiori. Se questa misurazione risulta anormale, viene eseguito un ecocardiogramma per ricercare un’eventuale anomalia cardiaca prima di lasciare il reparto di maternità.
Imaging delle cardiopatie congenite
risonanza magnetica
La risonanza magnetica rappresenta un importante contributo alla valutazione delle cardiopatie congenite, in particolare e soprattutto in età adulta, quando le prestazioni dell’ecocardiografia sono meno buone.
La risonanza magnetica fornisce informazioni aggiuntive essenziali per la competenza, il monitoraggio, la diagnosi, il trattamento e le decisioni terapeutiche.
La risonanza magnetica permette:
- Valutazione anatomica delle cavità e dei vasi
- Valutazione funzionale: della funzione miocardica
- Analisi dei flussi e delle portate
- La ricerca della fibrosi miocardica
- Test per l’ischemia utilizzando il test dell’adenosina
La sua attuazione è limitata nei bambini a causa della necessità di sedazione.
Le principali indicazioni riguardano ventricoli destri sistemici (doppia discordanza, trasposizione dei grandi vasi con switch atriale), ventricoli destri con sovraccarico di volume (tetralogia di Fallot), ventricoli singoli, malattia polmonare, aortica, tricuspide, mitralica, patologie aortiche.
Tomografia computerizzata
L’angio TAC è un imaging complementare che consente in particolare la visualizzazione dei vasi:
- arterie polmonari
- vene polmonari
- arterie coronarie
- aorta
- fistole veno-venose e artero-venose
La sua realizzazione è limitata dall’irradiazione e dall’iniezione di un mezzo di contrasto potenzialmente nefrotossico e allergenico.
Cardiopatie congenite in età adulta
- Oltre il 90% dei bambini affetti da cardiopatie congenite (CHD) sopravvive fino all’età adulta·
- Il numero di adulti affetti da CC è ora maggiore del numero di bambini.
- La maggior parte di questi pazienti non può essere considerata guarita e la loro cura è un processo che dura tutta la vita: dalla riparazione durante l’infanzia, al passaggio all’età adulta, al desiderio di gravidanza e alla gestione delle complicanze tardive specifiche di ciascun CC.
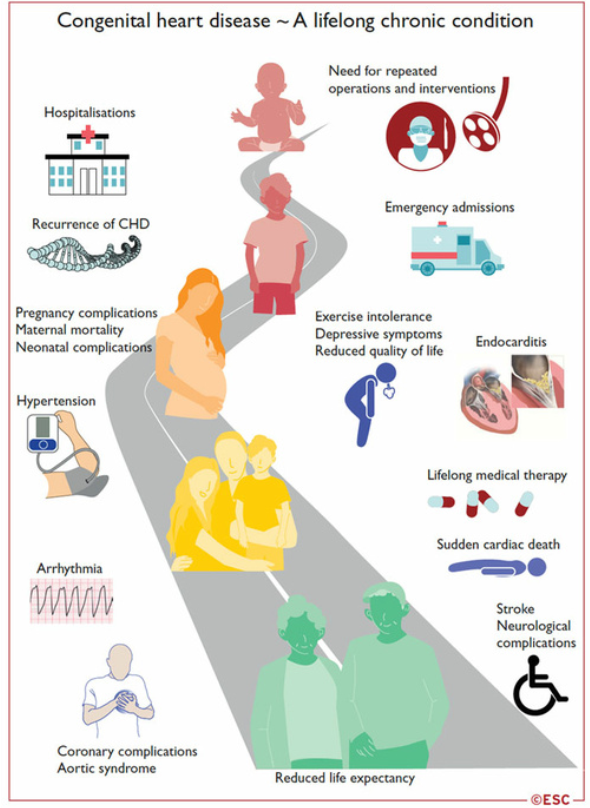
Transizione all’età adulta
Il passaggio alla fase adulta è importante e va anticipato e sostenuto, affinché il paziente possa ricevere tutte le informazioni sulla sua cardiopatia, sulla sua storia medica, sulle sue cure, sul suo follow-up e su tutte le problematiche legate alla sua cardiopatia.
Con questo obiettivo sono stati sviluppati e implementati programmi di educazione terapeutica nei centri di riferimento.
Al CCM: il passaggio avviene in modo naturale attraverso un’organizzazione basata su un’équipe medico-chirurgica che cura gli stessi pazienti dal feto all’età adulta.
Complicanze delle cardiopatie congenite in età adulta.
- Intervento di revisione: cardiopatia valvolare, dilatazione aortica, shunt residuo
- Disfunzione miocardica: ventricolo sinistro, ventricolo singolo, ventricolo destro sistemico
- Malattia coronarica
- Insufficienza cardiaca
- Aritmie
- Endocardite infettiva
- Ipertensione
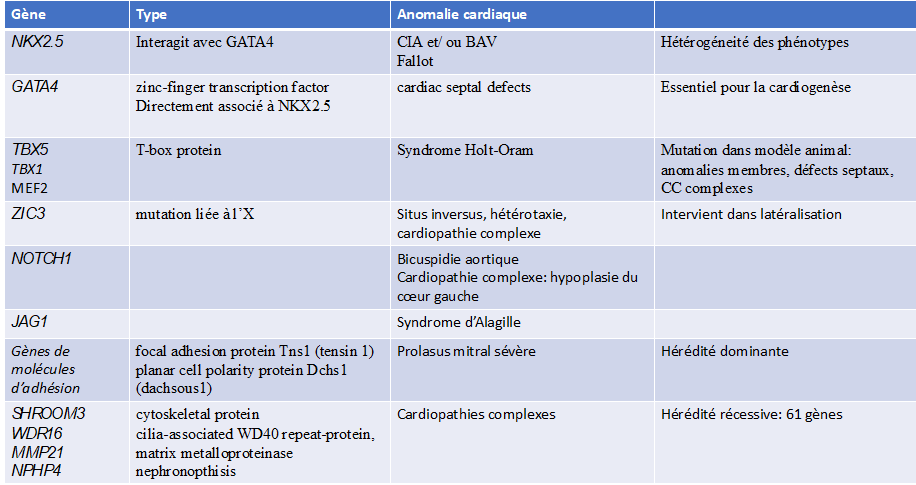
Trasmissione e genetica delle cardiopatie congenite
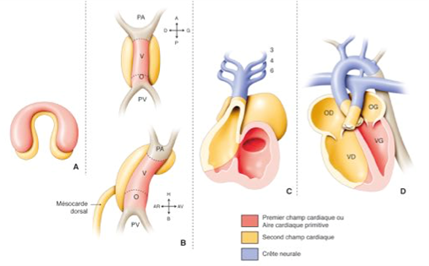
Embriologia del cuore normale: la formazione del cuore avviene durante le prime 8 settimane di vita fetale, secondo più fasi complesse successive a partire dal tubo cardiaco primitivo, con fenomeni di torsione e rotazione e partizionamento:
- Tubo cardiaco primario
- Loop: posizione delle cavità atri/ventricolari
- Formazione delle valvole atrioventricolari
- Divisione dei padiglioni auricolari
- Suddivisione dei ventricoli
- Suddivisione delle navi
Le cardiopatie congenite comportano un rischio di trasmissione ereditaria attraverso un meccanismo multifattoriale, sia genetico che ambientale.
Nella maggior parte dei casi, ovvero nel 72%, non è identificabile alcuna eziologia genetica.Fattori genetici e ambientali: rappresentano dal 20% al 30% dei casi
- Fattori ambientali: 2%
Malattie materne: diabete, rosolia, lupus eritematoso sistemico
Consumo materno di trattamenti quali: litio, isotretinoina, anticonvulsivanti o alcol e tabacco.
Età materna: questo è un fattore di rischio per la sindrome di Down, che può portare a difetti cardiaci.
- Anomalie cromosomiche (aneuploidie): 10%
trisomia 21 o sindrome di Down, trisomia 18, trisomia 13, monosomia X o sindrome di Turner.
- Delezioni subcromosomiche (microdelezioni), duplicazioni subcromosomiche, mutazionimonogenici:
sindromi congenite che colpiscono più organi
Sindrome di Di George (microdelezione 22q11.2)
Sindrome di Williams-Beuren (microdelezione 7p11.23)
Difetti di un singolo gene: mutazioni nella fibrillina-1 (sindrome di Marfan),
TXB5 (sindrome di Holt-Oram)
PTPN11 (sindrome di Noonan)
- Mutazione puntiforme autosomica dominante de novo: 8%
- Mutazione puntiforme ereditaria autosomica recessiva: 2%
Il rischio di recidiva di una cardiopatia congenita in una famiglia dipende dalla causa: trascurabile nelle mutazioni de novo, dal 2 al 5% nelle cardiopatie congenite multifattoriali non sindromiche e 50% quando è coinvolta una mutazione autosomica dominante.
È importante identificare i fattori genetici per valutare il rischio di recidiva e guidare la consulenza genetica preconcezionale.