Avec 11 796 kilomètres parcourus, le Centre Cardio-Thoracique a remporté la première place de la No Finish Line 2024,au classement général par équipe !
L’enthousiasme pour la No Finish Line 2024 ne faibli pas dans l’équipe, et nous sommes fiers d’avoir porté les couleurs du CCM !
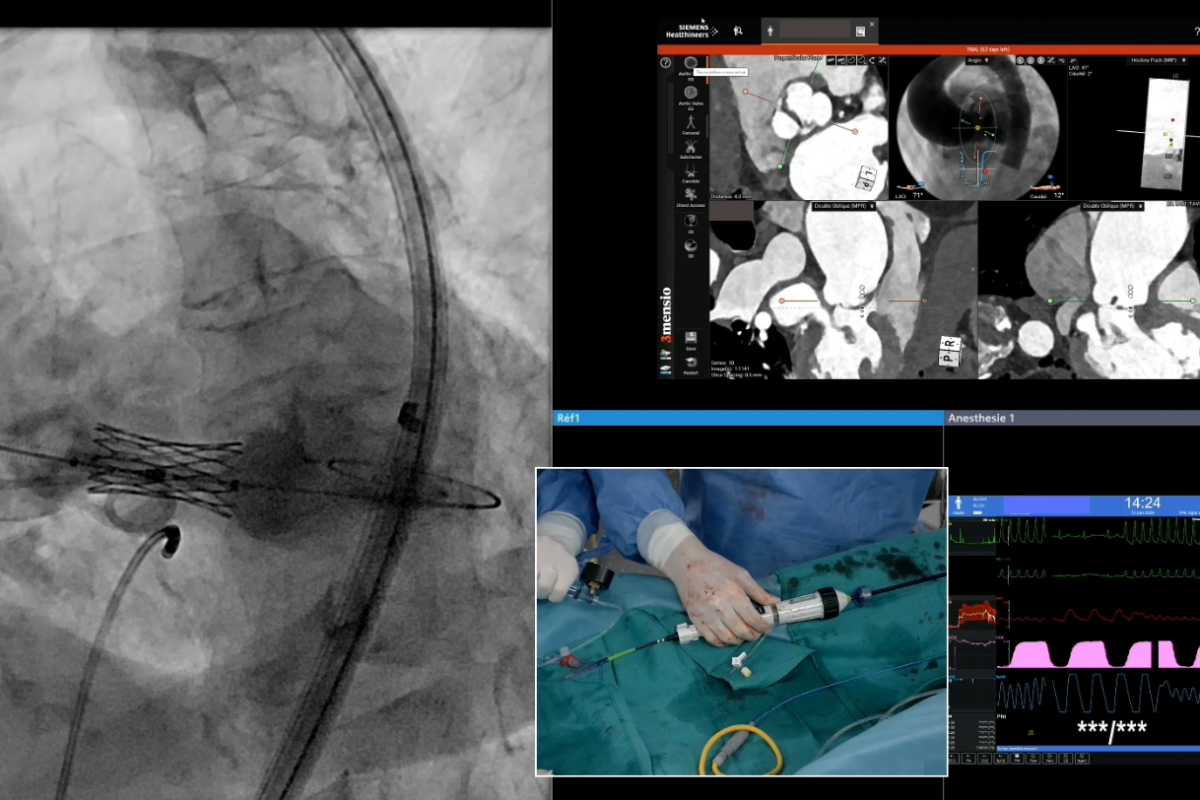
Il CCM è stato selezionato per eseguire dei live cases alla 9 edizione del Multi Level CTO, tenutosi a Nizza dal 27 al 29 giugno.
Durante i 3 giorni di questo congresso internazionale, dedicato esclusivamente al trattamento delle occlusioni coronariche totali croniche, sono stati trattati 4 live cases presso il CCM, consentendo ai cardiologi interventisti di seguire dal vivo casi reali di complessità crescente. I casi sono stati eseguiti dall’équipe di cardiologi interventisti del Centro, accompagnati da medici ospiti.
Il trattamento delle CTO mediante ricanalizzazione delle occlusioni coronariche totali croniche è diventato una procedura incontornabile, una specificità della cardiologia interventistica in forte espansione e in continua evoluzione. La padronanza delle tecniche di CTO è oggi essenziale per la gestione dei pazienti coronarici complessi.
Il capitano del Roca Team Yakuba Ouattara si è sempre impegnato ad aiutare i bambini malati e svantaggiati e da tre anni è il patrocinatore della No Finish Line, la più grande corsa di beneficenza di Monaco.
È venuto a trovare Aziz, 7 anni, che è stato operato dall’équipe di cardiochirurgia pediatrica del Centro cardio-toracico di Monaco, molto impegnato in attività umanitarie, dalla sua creazione. Il piccolo ivoriano soffriva di una grave malformazione cardiaca che non poteva essere curata nel suo Paese.
L’operazione è stata resa possibile grazie al coinvolgimento e alla generosità di Children & Future, del Dipartimento della Cooperazione Internazionale, del MCH, della Croce Rossa monegasca, di Aviation sans Frontières e delle famiglie ospitanti di Aziz.
Dare una seconda vita ai bambini malati dei Paesi in via di sviluppo è un impegno forte del Principato che mobilita questo sportivo dal cuore grande !
Il team di cardiologia interventistica del Centro ha ospitato le giornate REVES il 13 e 14 giugno. Organizzate intorno a 10 live cases eseguiti presso il Centro Cardio-Toracico di Monaco e commentati in diretta dagli operatori, il tema di queste giornate è stato il trattamento endovascolare delle valvulopatie e delle malattie strutturali. Più di 80 cardiologi interventisti provenienti da tutta la Francia, tra cui alcuni dei più eminenti, hanno assistito ai live cases e condiviso le loro esperienze. Le presentazioni dal vivo e i casi clinici hanno dato l’opportunità di discutere le strategie applicate, gli sviluppi delle tecniche nella pratica quotidiana e la gestione delle complicanze.
Ospitando questo evento e presentando casi clinici spesso complessi, il Centro Cardio-Toracico di Monaco perfeziona una delle sue attività principali, la cardiologia interventistica, e rafforza la sua posizione di Centro di riferimento e di eccellenza.
Pr. Sylvie DI FILIPPO Centre Cardio-Thoracique de Monaco. Cardiologia congenita pediatrica e adulti, cardiologia fetale – Professore delle Università.
I difetti cardiaci congeniti rappresentano le anomalie congenite d’organo più comuni. La loro incidenza nella popolazione in generale è di 14 feti ogni 1000 e 8 nati vivi ogni 1000. Le cardiopatie congenite comprendono malformazioni multiple, di complessità anatomo-funzionale variabile e prognosi eterogenea.
Queste malformazioni vengono classificate in base alla loro gravità e complessità in:
Semplici, moderate e severe
Riparabili e non riparabili
Biventricolari e univentricolari
Cianogene e non cianogene
Cardiopatie minori
Senza necessità di intervento
Cardiopatie moderate
Intervento indicato
Cardiopatie reparabili
Cardiopatie maggiori
Cardiopatie che necessitano un trattamento ed un intervento
Riparabile con degli effetti postumi o non reparabili
Valvola aortica bicuspide
Piccola comunicazione interatriale
Piccolo difetto del setto ventricolare
Piccolo dotto arterioso
Minore anomalia della valvola mitrale
stenosi polmonare di grado lieve
Anomalia delle vene polmonari
Anomalie coronariche
Stenosi aortica
Canale atrioventricolare
Ampio difetto interatriale (DIA)
Ampio difetto interventricolare (DIV)
Grande dotto arterioso
Coartazione aortica
Costrizione polmonare stretta
Malattia di Marfan e aneurisma aortico
Anomalia di Ebstein
Tetralogia di Fallot
Trasposizione dei grandi vasi dopo la riparazione
Malattia cardiaca con ipertensione polmonare fissa (Eisenmenger)
Cardiopatia cianogena non operata o con palliazione
Interruzione dell’arco aortico
Ventricolo destro a doppia uscita
Ventricoli singoli
Atresia polmonare
Truncus arteriosus
Circolazione univentricolare di Fontan
Trasposizione dei grandi vasi con ventricolo destro sotto l’aorta
Cardiopatie complesse con lesioni multiple
Classificazione anatomo-fisiopatologica delle cardiopatie congenite
Shunt sinistro-destro
Shunt destro-sinistro e ostacoli destri
Ostacoli lasciati
Anomalie coronariche
Anomalie del ritmo cardiaco
Malattie cardiache complesse
Shunt: comunicazione anormale naturale o creata tra camere o vasi cardiaci.
Cianosi: colorazione bluastra della pelle e delle mucose dovuta a un calo di ossigeno nel sangue (o ipossia).
Screening prenatale
I difetti cardiaci congeniti sono ben tollerati durante la vita intrauterina e non impediscono il normale sviluppo del feto.
L’adattamento alla vita extrauterina, che comporta un calo della pressione polmonare e la chiusura del dotto arterioso e del forame ovale provoca lo scompenso delle malformazioni cardiache la cui vitalità dipende dalla permeabilità di questi shunt.
La diagnosi prenatale della cardiopatia mediante ecocardiografia e Doppler consente di anticipare e prevenire il suo scompenso, guidando l’immediata condotta terapeutica postnatale. Il parto viene programmato e pianificato dall’équipe gineco-ostetrica e neonatologica.
Nella maggior parte dei casi, l’induzione prematura non è necessaria. Solo i casi di insufficienza cardiaca fetale possono giustificarlo (conseguenza di un ritmo cardiaco anomalo, sia troppo lento = bradicardia fetale, sia troppo veloce = tachicardia fetale incontrollata).
Lo screening prenatale consente la diagnosi di cardiopatie con una performance che varia dal 40% al 90% a seconda del tipo di cardiopatia
Permette di stabilire una diagnosi, monitorare i progressi prenatali, pianificare l’assistenza postnatale e stabilire la prognosi
Il parto così programmato aiuta a prevenire lo scompenso delle malattie cardiache dopo la nascita
La somministrazione della Prostaglandina E1 è un trattamento che mantiene aperto il dotto arterioso alla nascita, mantenendo la circolazione e/o l’ossigenazione nei neonati con malattie cardiache a rischio di scompenso acuto.
Le malattie cardiache a rischio di scompenso neonatale che comporta un rischio letale includono:
• Ostacoli diritti che limitano il flusso polmonare
• Ostacoli sinistri che limitano il flusso aortico
• Anomalie di mescolamento che dipendono dalla permeabilità del forame ovale.
Diagnosi e sintomi clinici
Neonato
Cianosi, insufficienza cardiaca
Cardiopatia shunt-dipendente
Sintomi
Difficoltà di alimentazione
Scarsa crescita o aumento delle infezioni broncopolmonari
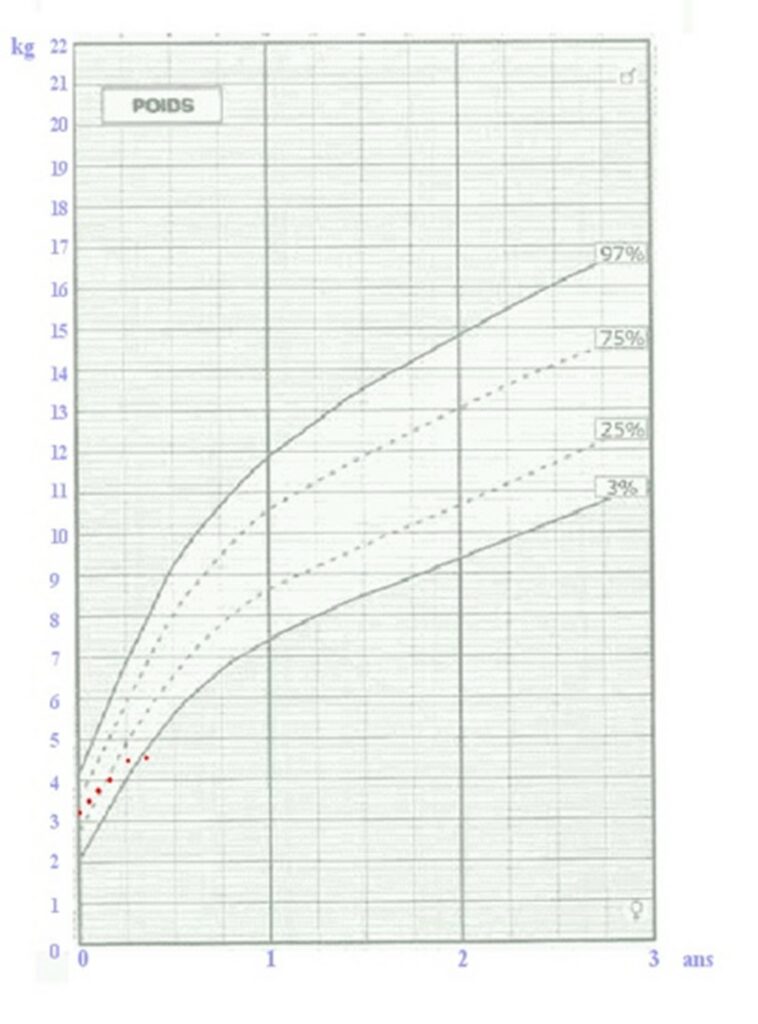
Auscultazione: rilevamento di un soffioCurva di crescita del peso di un neonato: stagnazione del peso (punti rossi)
Effettuare un’ecografia cardiaca e un Doppler: l’ecografia cardiaca è un esame semplice e non invasivo che può essere utilizzato per diagnosticare difetti cardiaci a qualsiasi età.
Neonatologia: screening per le cardiopatie congenite
Prenatale: la cardiopatia rilevata prima della nascita deve essere confermata dopo la nascita mediante ecografia cardiaca nel neonato.
Postnatale: non tutte le malattie cardiache vengono rilevate prima della nascita e l’esame di tutti i neonati nel reparto maternità resta molto importante per cercare un’eventuale anomalia cardiaca non precedentemente diagnosticata cosi da riconoscerla prima del ritorno a casa.
L’esame clinico deve ricercare:
Palpazione dei polsi femorali
La presenza di un soffio all’ausculazione
Il comportamento alimentare del neonato
Saturazione di ossigeno o satO2: lo screening sistematico per le cardiopatie neonatali viene attuato nei reparti maternità con la misurazione della saturazione di ossigeno negli arti superiori e inferiori, il cui tasso normale è > 97% con una differenza inferiore al 3% tra gli arti superiori e arti inferiori. Se questa misurazione risulta anormale, viene eseguito un ecocardiogramma per ricercare un’eventuale anomalia cardiaca prima di lasciare il reparto di maternità.
Sensore di saturazione del saturometro
Imaging delle cardiopatie congenite
risonanza magnetica
La risonanza magnetica rappresenta un importante contributo alla valutazione delle cardiopatie congenite, in particolare e soprattutto in età adulta, quando le prestazioni dell’ecocardiografia sono meno buone.
La risonanza magnetica fornisce informazioni aggiuntive essenziali per la competenza, il monitoraggio, la diagnosi, il trattamento e le decisioni terapeutiche.
La risonanza magnetica permette:
Valutazione anatomica delle cavità e dei vasi
Valutazione funzionale: della funzione miocardica
Analisi dei flussi e delle portate
La ricerca della fibrosi miocardica
Test per l’ischemia utilizzando il test dell’adenosina
La sua attuazione è limitata nei bambini a causa della necessità di sedazione.
Le principali indicazioni riguardano ventricoli destri sistemici (doppia discordanza, trasposizione dei grandi vasi con switch atriale), ventricoli destri con sovraccarico di volume (tetralogia di Fallot), ventricoli singoli, malattia polmonare, aortica, tricuspide, mitralica, patologie aortiche.
Tomografia computerizzata
L’angio TAC è un imaging complementare che consente in particolare la visualizzazione dei vasi:
arterie polmonari
vene polmonari
arterie coronarie
aorta
fistole veno-venose e artero-venose
La sua realizzazione è limitata dall’irradiazione e dall’iniezione di un mezzo di contrasto potenzialmente nefrotossico e allergenico.
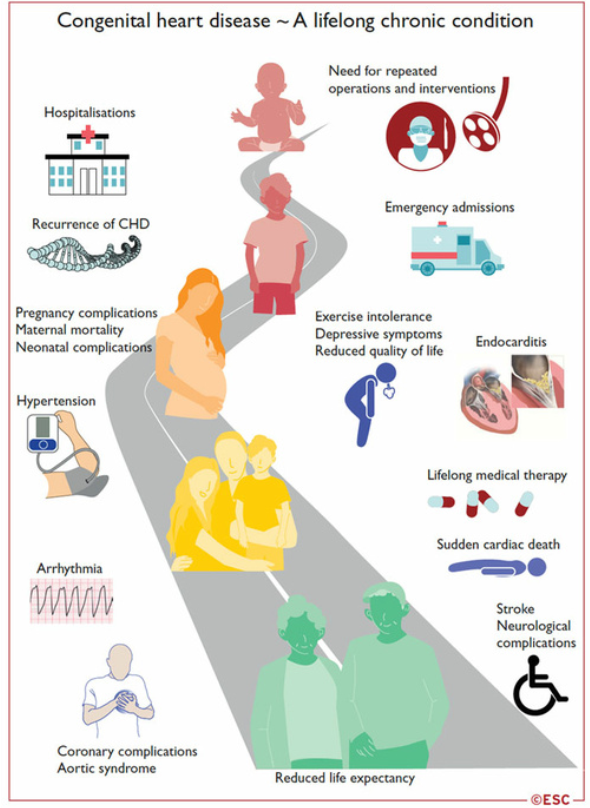
Cardiopatie congenite in età adulta
Oltre il 90% dei bambini affetti da cardiopatie congenite (CHD) sopravvive fino all’età adulta·
Il numero di adulti affetti da CC è ora maggiore del numero di bambini.
La maggior parte di questi pazienti non può essere considerata guarita e la loro cura è un processo che dura tutta la vita: dalla riparazione durante l’infanzia, al passaggio all’età adulta, al desiderio di gravidanza e alla gestione delle complicanze tardive specifiche di ciascun CC.
Raccomandazioni ESC 2020: gestione delle cardiopatie congenite in età adulta.
Transizione all’età adulta
Il passaggio alla fase adulta è importante e va anticipato e sostenuto, affinché il paziente possa ricevere tutte le informazioni sulla sua cardiopatia, sulla sua storia medica, sulle sue cure, sul suo follow-up e su tutte le problematiche legate alla sua cardiopatia.
Con questo obiettivo sono stati sviluppati e implementati programmi di educazione terapeutica nei centri di riferimento.
Al CCM: il passaggio avviene in modo naturale attraverso un’organizzazione basata su un’équipe medico-chirurgica che cura gli stessi pazienti dal feto all’età adulta.
Complicanze delle cardiopatie congenite in età adulta.
Intervento di revisione: cardiopatia valvolare, dilatazione aortica, shunt residuo
Trasmissione e genetica delle cardiopatie congenite
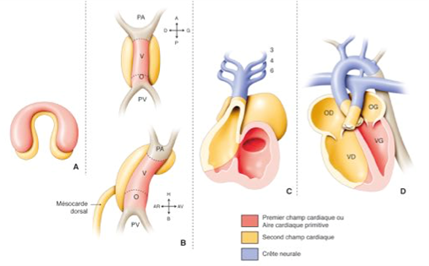
Embriologia del cuore normale: la formazione del cuore avviene durante le prime 8 settimane di vita fetale, secondo più fasi complesse successive a partire dal tubo cardiaco primitivo, con fenomeni di torsione e rotazione e partizionamento:
Tubo cardiaco primario
Loop: posizione delle cavità atri/ventricolari
Formazione delle valvole atrioventricolari
Divisione dei padiglioni auricolari
Suddivisione dei ventricoli
Suddivisione delle navi
Le cardiopatie congenite comportano un rischio di trasmissione ereditaria attraverso un meccanismo multifattoriale, sia genetico che ambientale.
Nella maggior parte dei casi, ovvero nel 72%, non è identificabile alcuna eziologia genetica.Fattori genetici e ambientali: rappresentano dal 20% al 30% dei casi
Consumo materno di trattamenti quali: litio, isotretinoina, anticonvulsivanti o alcol e tabacco.
Età materna: questo è un fattore di rischio per la sindrome di Down, che può portare a difetti cardiaci.
Anomalie cromosomiche (aneuploidie): 10%
trisomia 21 o sindrome di Down, trisomia 18, trisomia 13, monosomia X o sindrome di Turner.
Delezioni subcromosomiche (microdelezioni), duplicazioni subcromosomiche, mutazionimonogenici:
sindromi congenite che colpiscono più organi
Sindrome di Di George (microdelezione 22q11.2)
Sindrome di Williams-Beuren (microdelezione 7p11.23)
Difetti di un singolo gene: mutazioni nella fibrillina-1 (sindrome di Marfan),
TXB5 (sindrome di Holt-Oram)
PTPN11 (sindrome di Noonan)
Mutazione puntiforme autosomica dominante de novo: 8%
Il rischio di recidiva di una cardiopatia congenita in una famiglia dipende dalla causa: trascurabile nelle mutazioni de novo, dal 2 al 5% nelle cardiopatie congenite multifattoriali non sindromiche e 50% quando è coinvolta una mutazione autosomica dominante.
È importante identificare i fattori genetici per valutare il rischio di recidiva e guidare la consulenza genetica preconcezionale.
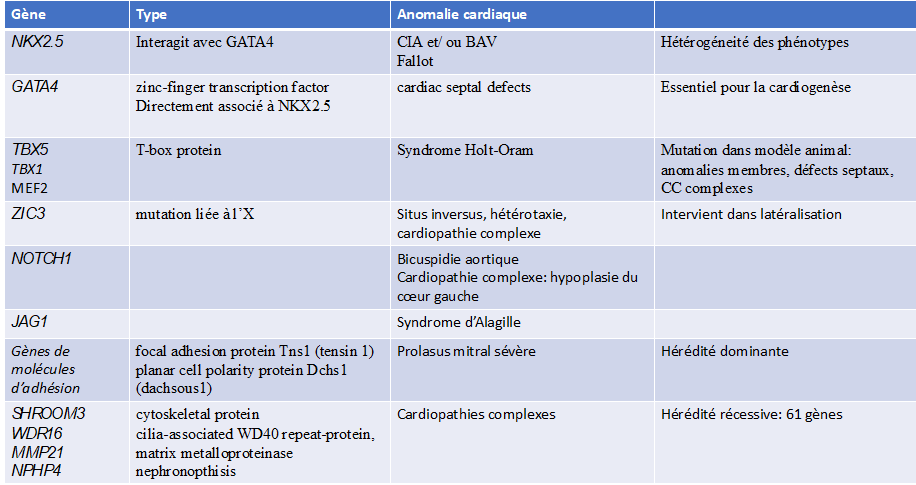
Tabella: esempi di mutazioni genetiche con malattie cardiache associate
Contatto +377 92 16 80 00 Le urgenze cardiovascolari e toraciche sono accettate senza restrizioni, 24 ore su 24, 7 giorni su 7, compresi domenica e festivi.