Pr. Sylvie DI FILIPPO
Monaco Cardiothoracic Center.
Pediatric and adult congenital cardiology & fetal cardiology.
University professor, hospital practitioner.
Congenital heart disease is the most common congenital organ anomaly. Their incidence in the general population is 14 foetuses per 1000 and 8 births per 1000.
Congenital heart disease includes multiple malformations of varying anatomical and functional complexity and with heterogeneous prognoses.
These malformations are classified according to their severity and complexity into:
- Minor, moderate and major
- Repairable and non-repairable
- Biventricular and univentricular
- Cyanogenic and non-cyanogenic
Minor heart disease
|
Moderate heart disease
|
Severe heart disease
|
|
|
|
Anatomo-physiopathological classification of congenital heart disease
- Left-right shunts
- Right-to-left shunts and right obstructions
- Left obstructions
- Coronary anomalies
- Rhythmological abnormalities
- Complex heart disease
Shunt: natural or created abnormal communication between heart chambers or vessels.
Cyanosis: blue discolouration of the skin and mucous membranes due to a drop in oxygen in the blood (or hypoxia).
Antenatal screening
- Congenital heart disease is well tolerated during intrauterine life and does not prevent normal development of the foetus.
- Adaptation to extra-uterine life, which involves a fall in pulmonary pressures and closure of the ductus arteriosus and foramen ovale, is the cause of decompensation of cardiac malformations, the viability of which depends on the permeability of these shunts.
- Antenatal diagnosis of heart disease by echocardiography and Doppler enables decompensation to be anticipated and prevented, by guiding immediate post-natal therapeutic management. The birth is programmed and planned by the gynaeco-obstetric and neonatology team.
- In the majority of cases, pre-term induction is not necessary. Only cases of fetal heart failure may justify it (the consequence of an abnormal heart rhythm, either too slow = fetal bradycardia, or too fast = uncontrolled fetal tachycardia).
- Antenatal screening enables heart disease to be diagnosed, with results ranging from 40% to 90% depending on the type of heart disease.
- It enables a diagnosis to be made, the course of the disease to be monitored antenatally, postnatal care to be planned and the prognosis to be established.
- Birth planned in this way helps prevent decompensation of the heart disease after birth
- The prostaglandin E1 administered is a treatment that keeps the ductus arteriosus open at birth, thereby maintaining circulation and/or oxygenation in newborns with heart disease at risk of acute decompensation.
- Heart diseases at risk of neonatal decompensation entail a lethal risk, including::
– Right-sided obstructions that limit pulmonary flow
– Left-sided obstructions that limit aortic flow
– Mixing anomalies that depend on the patency of the foramen ovale
Diagnosis and clinical symptoms
Newborn
- Cyanosis, heart failure
- Shunt-dependent heart disease
Symptoms
- Feeding difficulties
- Poor growth or weight stagnation
- Frequent bronchopulmonary infections
- Dyspnoea on exertion
- Feeling unwell, syncope
- Chest pain
Asymptomatic
- Breathing
- ECG abnormality discovered by chance
- Abnormalities on clinical examination: femoral pulses, high blood pressure
Cardiac and Doppler ultrasound: cardiac ultrasound is a simple, non-invasive examination that can be used to diagnose cardiac malformations at any age.
Neonatology
Screening for congenital heart disease
Antenatal
heart disease detected antenatally must be confirmed postnatally by a cardiac ultrasound scan of the newborn.
Postnatally
not all heart disease is detected before birth, and it is still very important to examine all newborn babies in the maternity unit to look for any heart abnormality that may have gone unrecognised, and to recognise it before the baby is discharged home.
The clinical examination should look for :
- palpation of the femoral pulses
- The presence of a murmur on ausculation
- The newborn’s feeding behaviour
- Oxygen saturation or satO2: systematic screening for heart disease in newborns is carried out in maternity units by measuring oxygen saturation in the upper and lower limbs. The normal level is > 97%, with a difference of less than 3% between the upper and lower limbs. If this measurement is abnormal, an echocardiogram is performed to check for any cardiac abnormality before the baby is discharged from the maternity hospital.
Imaging congenital heart disease
IRM
Magnetic resonance imaging makes a major contribution to the assessment of congenital heart disease, particularly in adulthood, when the performance of echocardiography is less good.
MRI provides additional information that is essential for assessment, follow-up, diagnosis, management and therapeutic decisions.
It allows :
- Anatomical assessment of cavities and vessels
- Functional assessment: of myocardial function
- Analysis of flow and output
- Search for myocardial fibrosis
- Testing for ischaemia using the adenosine test
Its use is limited in children because of the need for sedation
The main indications are systemic right ventricles (double mismatch, transposition of the great vessels with atrial switch), right ventricles with volume overload (tetralogy of Fallot), single ventricles, pulmonary, aortic, tricuspid and mitral valve disease, aortic pathologies, etc.
SCANNER
The angio-scanner is a complementary imaging technique which allows the vessels to be viewed in particular:
- pulmonary arteries
- pulmonary veins
- coronary arteries
- aorta
- veno-venous and arteriovenous fistulas
Its use is limited by irradiation and injection of potentially nephrotoxic and allergenic contrast medium.
Congenital heart disease in adulthood
- Over 90% of children with congenital heart disease (CHD) survive into adulthood.
- The number of adults with CHD is now greater than the number of children.
- Most of these patients cannot be considered cured, and their management is a lifelong process: from repair in childhood, to the transition to adulthood, to the desire for pregnancy, and the management of late complications specific to each CC.
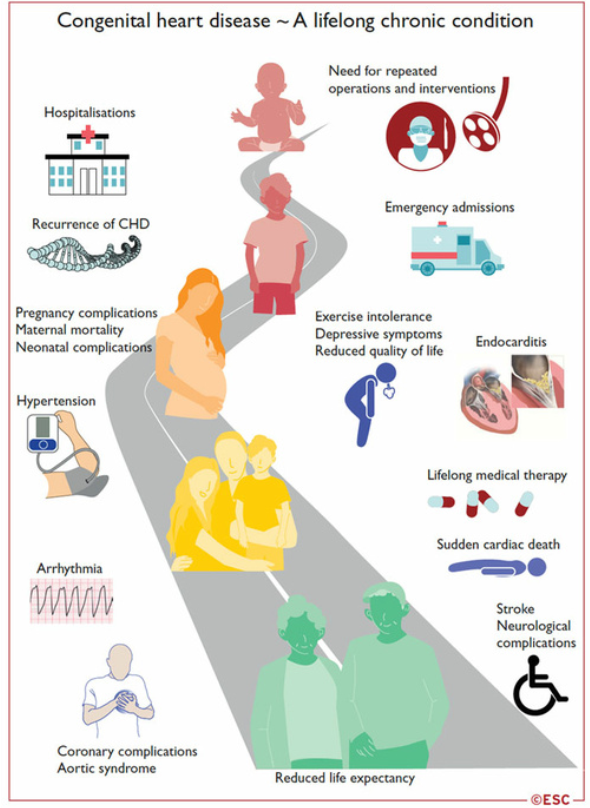
Transition to adulthood
The transition to adulthood is an important phase that needs to be anticipated and supported, so that patients can receive all the information they need about their heart disease, their medical history, their treatment, their follow-up and all the problems associated with their heart disease.
To this end, therapeutic education programmes have been developed and set up in the referral centres.
At the CCM: the transition is made naturally by an organisation based on a medical-surgical team caring for the same patients from foetus to adulthood.
Complications of congenital heart disease in adulthood:
- Surgical revision: valvulopathy, dilated aorta, residual shunt
- Myocardial dysfunction: left ventricle, single ventricle, systemic right ventricle
- Coronary artery disease
- Heart failure
- Arrhythmias
- Infectious endocarditis
- Arterial hypertension
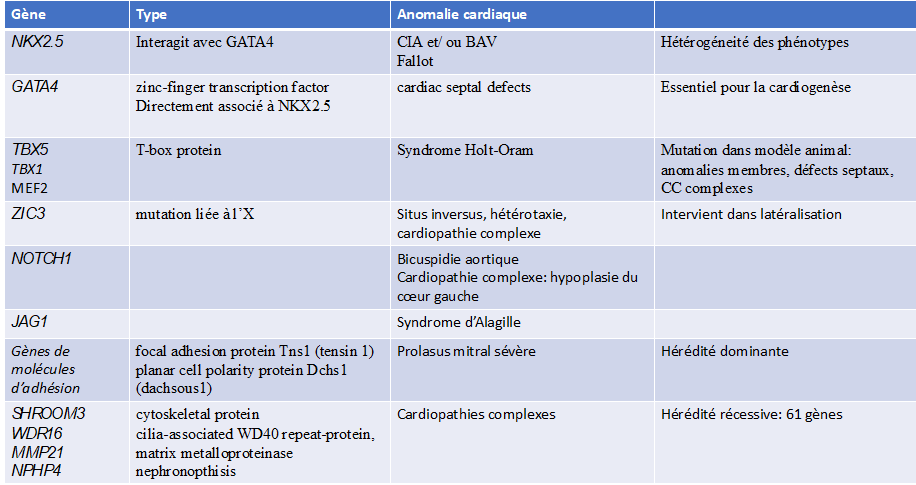
Transmission and genetics of congenital heart disease
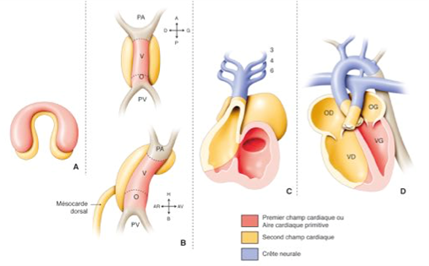
Embryology of the normal heart: the heart is formed during the first 8 weeks of foetal life, in several successive complex stages starting from the primitive cardiac tube, with phenomena of torsion and rotation and partitioning:
- Primitive heart tube
- Loop: position of the atria/ventricles cavities
- Formation of the atrioventricular valves
- Partitioning of the atria
- Partitioning of the ventricles
- Partitioning of the vessels
 Congenital heart disease is inherited through a multifactorial mechanism involving both genetic and environmental factors.
Congenital heart disease is inherited through a multifactorial mechanism involving both genetic and environmental factors.
In the majority of cases (72%), no genetic aetiology is identifiable.
- Genetic and environmental factors: account for 20% to 30% of cases
- Environmental factors: 2% of cases
Maternal illnesses: diabetes, rubella, systemic lupus erythematosus.
Maternal use of treatments such as lithium, isotretinoin and anticonvulsants.
Maternal age: this is a risk factor for trisomy 21, which can lead to heart malformations.
- Chromosomal abnormalities (aneuploidies): 10%.
trisomy 21 or Down syndrome, trisomy 18, trisomy 13, monosomy X or Turner syndrome
- Subchromosomal deletions (microdeletions), subchromosomal duplications, single-gene mutations
monogenic: congenital syndromes affecting multiple organs
Di George syndrome (microdeletion 22q11.2)
Williams-Beuren syndrome (microdeletion 7p11.23)
Single gene defects: fibrillin-1 mutations (Marfan syndrome), TXB5 (Holt-Oram syndrome)
PTPN11 (Noonan syndrome).
- Autosomal dominant de novo point mutation: 8%.
- Autosomal recessive inherited point mutation: 2%.
The risk of recurrence of congenital heart disease in a family depends on the cause: negligible in de novo mutations, 2 to 5% in non-syndromic multifactorial congenital heart disease, and 50% when an autosomal dominant mutation is involved.
It is important to identify genetic factors in order to assess the risk of recurrence and to guide preconception genetic counselling.