Le 18 septembre, l’ensemble des professionnels, des partenaires, des bénévoles et de l’effectif de l’AS Monaco Rugby s’est réuni au Centre Cardio-Thoracique de Monaco, pour la soirée de lancement de la saison en Fédéral 2. Au cours de ce moment très amical, les joueurs se sont rassemblés pour la photo officielle 2024/2025.
Monsieur Guy Nervo, Président Délégué du CCM, a rappelé que cet événement scellait, non seulement le partenariat du Centre avec l’AS Monaco Rugby depuis p)lus de 10 ans, mais également qu’il fédérait leurs valeurs communes : l’engagement collectif, l’esprit d’équipe, la cohésion, la complémentarité, la solidarité, le respect et la performance. Symbole de cette collaboration, Monsieur Thomas Riqué, Président de l’AS Monaco Rugby, a remis à Monsieur Guy Nervo un ancien maillot de l’équipe.
Tous les collaborateurs du CCM souhaitent aux équipes de l’AS Monaco Rugby beaucoup de succès pour la saison 2024/2025, avec pour objectif principal, celui de monter en Fédéral 1.
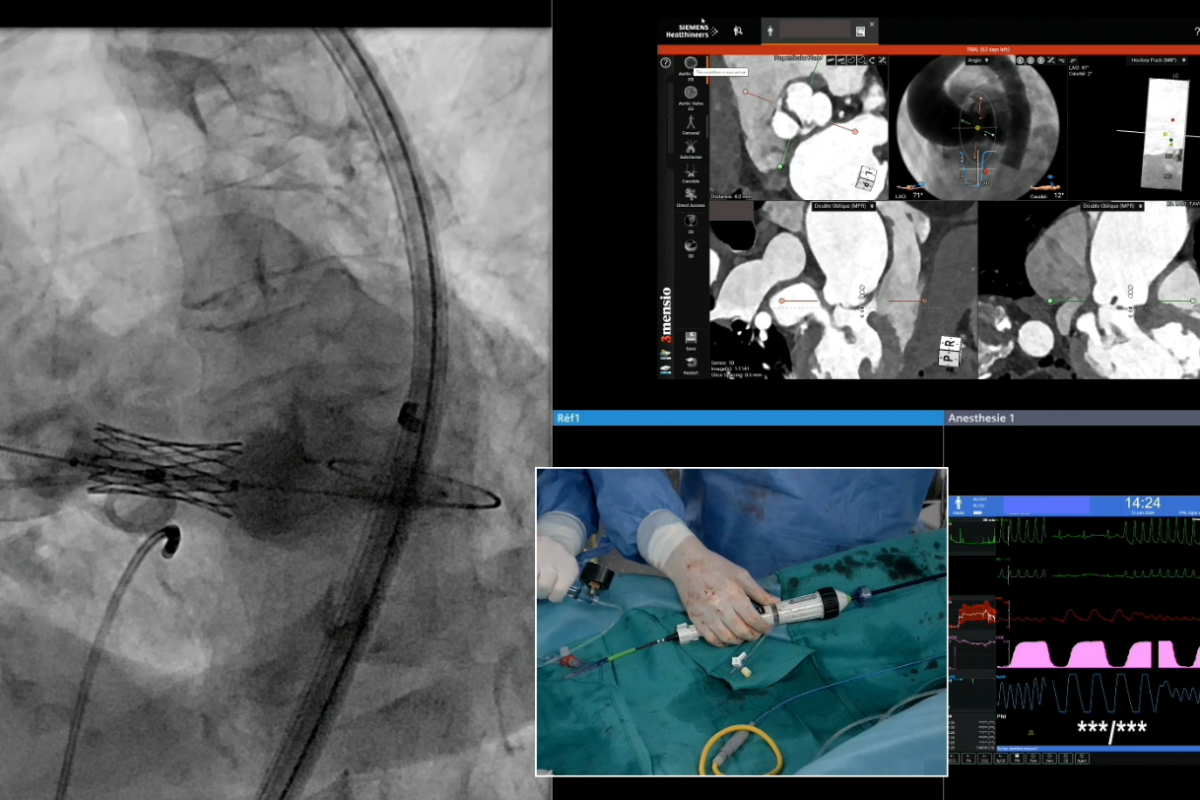
Le Centre Cardio-Thoracique de Monaco a été retenu pour la réalisation de live cases lors du 9e Multi Level CTO Course du 27 au 29 juin, qui s’est tenu à Nice.
Durant les 3 jours de ce congrès international, exclusivement dédié au traitement des occlusions coronariennes chroniques totales, 4 live cases ont été réalisés au CCM, permettant aux cardiologues interventionnels de suivre en direct des cas réels à la complexité croissante. Ils ont été effectués par l’équipe des cardiologues interventionnels du Centre accompagnés de praticiens invités.
Le traitement de la CTO par la re canalisation des occlusions coronariennes chroniques totales est devenu une procédure incontournable, une spécificité de la cardiologie interventionnelle en plein essor et en constante évolution. Maîtriser les techniques de CTO est aujourd’hui essentiel pour gérer des patients coronariens complexes.
Le capitaine de la Roca Team, Yakuba Ouattara, s’est toujours engagé auprès des enfants malades et démunis, notamment en étant depuis 3 ans le parrain de la No Finish Line, grande course caritative monégasque.
Il est ainsi venu voir le petit Aziz âgé de 7ans, qui a été opéré par l’équipe cardio-pédiatrique du Centre Cardio-Thoracique de Monaco, très impliqué dans les actions humanitaires depuis sa création. Ce petit Ivoirien souffrait d’une malformation cardiaque grave qui ne pouvait être traitée dans son pays d’origine.
Cette opération a pu être réalisée grâce à l’implication et à la générosité de Children & Future, la Direction de la Coopération Internationale, le MCH, la Croix Rouge Monégasque, Aviation sans Frontières et les familles d’accueil d’Aziz.
Offrir une seconde vie à des enfants malades issus de pays en voie développement est un engagement fort de la Principauté et mobilisateur ce sportif au grand cœur !
L’équipe de cardiologie interventionnelle du Centre a accueilli les Journées REVES les 13 et 14 juin. Organisées autour de 10 live cases réalisés au Centre Cardio-Thoracique de Monaco et commentés en direct par les opérateurs, ces journées avaient pour thématique le traitement endovasculaire des valvulopathies et maladies structurelles.
Plus de 80 cardiologues interventionnels de toute la France, dont les plus éminents, étaient présents pour assister aux live cases et partager leurs expériences. Autour des interventions en direct, de présentations de cas cliniques, ils ont ainsi pu échanger sur les stratégies appliquées, les évolutions des techniques dans la pratique quotidienne et la gestion des complications.
En accueillant ces Journées et y présentant des cas cliniques souvent complexes, le Centre Cardio-Thoracique de Monaco parfait une de ses activités majeures, la cardiologie interventionnelle et renforce ainsi son positionnement de Centre de Référence et d’Excellence.
Pr. Sylvie DI FILIPPO Centre Cardio-Thoracique de Monaco. Cardiologie congénitale pédiatrique et adulte & cardiologie fœtale. Professeur des Universités, praticien hospitalier.
Les cardiopathies congénitales représentent les anomalies d’organe congénitales les plus fréquentes. Leur incidence dans la population générale est de 14 fœtus pour 1000 et 8 naissances pour 1000. Elles incluent des malformations multiples, de complexité variable sur le plan anatomique et fonctionnel et de pronostic hétérogène.
Ces malformations sont classées en fonction de leur gravité et leur complexité en :
Mineures, modérées et majeures
Réparables et non réparables
Biventriculaires et univentriculaires
Cyanogènes et non cyanogènes
Cardiopathies mineures
Pas de nécessité d’intervention
Cardiopathies modérées
Intervention indiquée
Cardiopathies réparables
Cardiopathies sévères
Cardiopathies nécessitant traitement et intervention
Réparables avec séquelles ou non réparables
Bicuspidie valve aortique
Petite communication inter-auriculaire
Petite communication interventriculaire
Petit canal artériel
Anomalie mineure de la valve mitrale
Rétrécissement pulmonaire mineur
Anomalie des veines pulmonaires
Anomalies coronaires
Rétrécissement aortique
Canal atrioventriculaire
Large CIA
Large CIV
Large canal artériel
Coarctation aortique
Rétrécissement pulmonaire serré
Maladie de Marfan et anévrisme de l’aorte
Anomalie tricuspide d’Ebstein
Tétralogie de Fallot
Transposition des gros vaisseaux après réparation
Cardiopathies avec hypertension pulmonaire fixée (Eisenmenger)
Cardiopathies cyanogènes non opérée ou avec palliation
Interruption de l’arche aortique
Ventricule droit à double issue
Ventricules uniques
Atrésies pulmonaires
Tronc artériel commun
Circulation univentriculaire de Fontan
Transposition des gros vaisseaux avec ventricule droit sous aortique
Cardiopathies complexes avec lésions multiples
Classification anatomo-physiopathologiques des cardiopathies congénitales
Shunts gauche-droit
Shunts droit-gauche et obstacles droits
Obstacles gauches
Anomalies coronaires
Anomalies rythmologiques
Cardiopathies complexes
Shunt : communication anormale naturelle ou créée entre cavités cardiaques ou vaisseaux.
Cyanose : coloration bleue de la peau et des muqueuses dûe à une baisse d’oxygène dans le sang (ou hypoxie).
Le dépistage anténatal
Les cardiopathies congénitales sont bien tolérées pendant la vie intra-utérine et n’empêchent pas le développement normal du foetus.
L’adaptation à la vie extra-utérine qui suppose chute des pressions pulmonaires et fermeture du canal artériel et du foramen ovale, est cause de décompensation des malformations cardiaques dont la viabilité dépend de la perméabilité de ces shunts.
Le diagnostic anténatal de la cardiopathie par échocardiographie et Doppler, permet d’anticiper et prévenir sa décompensation, en guidant la conduite thérapeutique post-natale immédiate. La naissance est programmée et planifiée par l’équipe gynéco-obstétricale et néonatologiste.
Dans la majorité des cas, le déclenchement avant terme n’est pas nécessaire. Seuls les cas d’insuffisance cardiaque du fœtus peuvent le justifier (conséquence de rythme cardiaque anormal, soit trop lent= bradycardie foetale, soit trop rapide= tachycardie fœtale non contrôlée).
Le dépistage anténatal permet le diagnostic de la cardiopathie avec une performance variant de 40% à 90% selon le type de cardiopathie
Il permet d’établir un diagnostic, suivre l’évolution en anténatal, prévoir la prise en charge postnatale et établir le pronostic
La naissance ainsi programmée permet de prévenir la décompensation de la cardiopathie après la naissance
La prostaglandine E1 administrée est un traitement qui permet de garder ouvert le canal artériel à la naissance, ce qui maintient la circulation et/ou l’oxygénation chez le nouveau-né avec une cardiopathie à risque de décompensation aigue.
Les cardiopathies à risque de décompensation néonatale entrainent un risque létal :
• Les obstacles droits qui limitent le débit pulmonaire
• Les obstacles gauches qui limitent le débit aortique
• Les anomalies de mixing qui dépendent de la perméabilité du foramen ovale
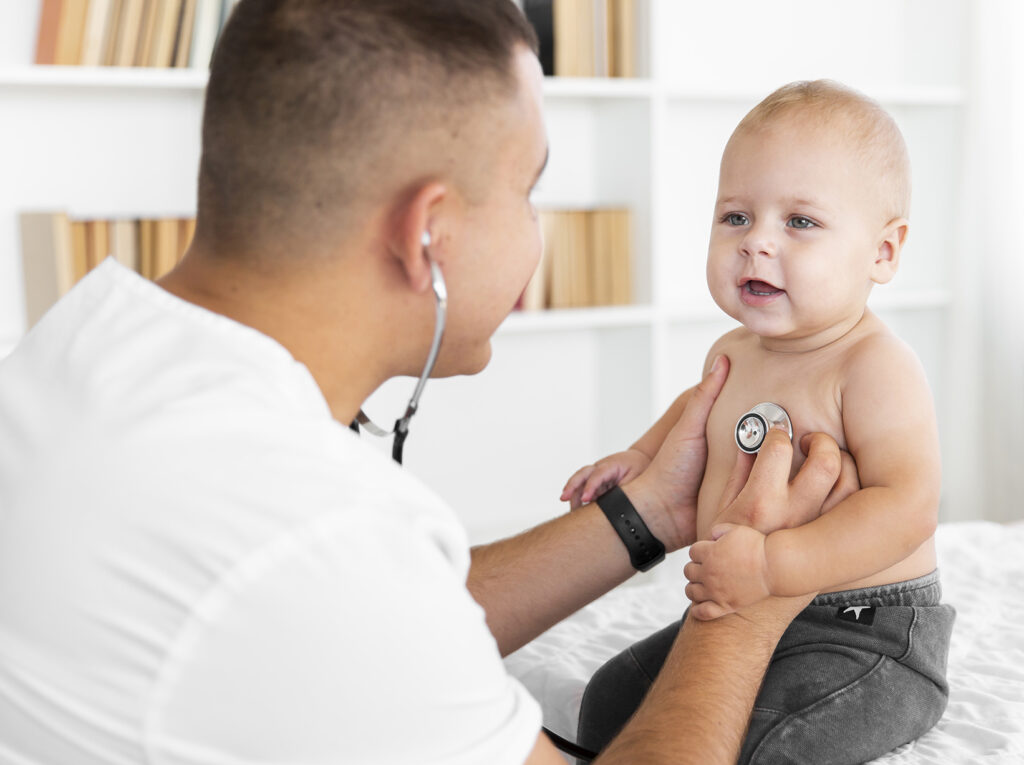
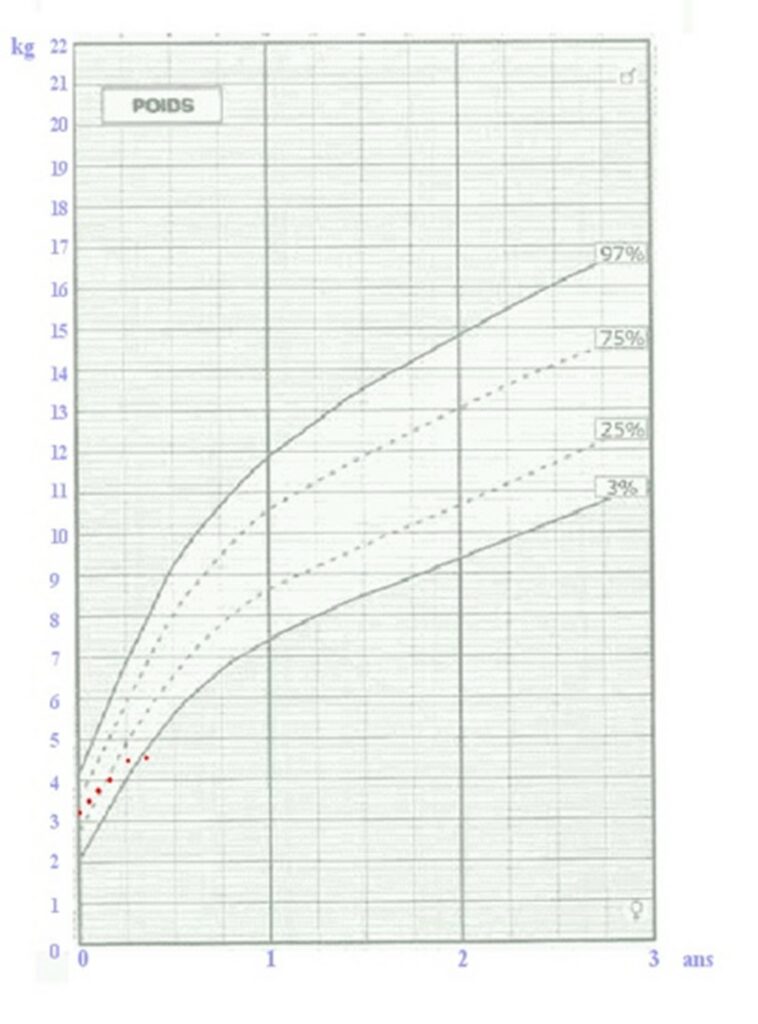
Auscultation : détection d’un souffleCourbe de croissance pondérale d’un nourrisson : stagnation du poids (points rouges)
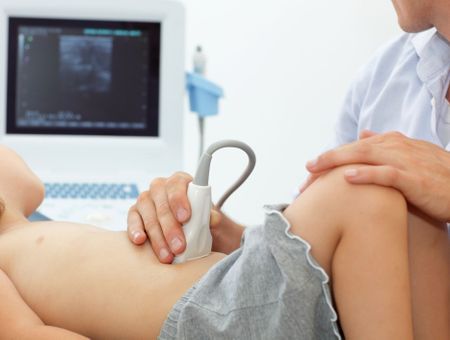
Faire une échographie cardiaque et Doppler : l’échographie cardiaque est un examen simple et non invasif qui permet de faire le diagnostic des malformations cardiaques à tout âge.
Néonatologie : Dépistage des cardiopathies congénitales
En anténatal : la cardiopathie dépistée en anténatal doit être confirmée en post natal par une échographie cardiaque chez le nouveau-né.
En postnatal : toutes les cardiopathies ne sont pas dépistées avant la naissance et l’examen de tous les nouveaux-nés à la maternité reste très important pour rechercher une éventuelle anomalie cardiaque qui serait méconnue et la reconnaître avant le retour à domicile.
L’examen clinique doit rechercher :
La palpation des pouls fémoraux
La présence d’un souffle à l’ausculation
Le comportement alimentaire du nouveau-né
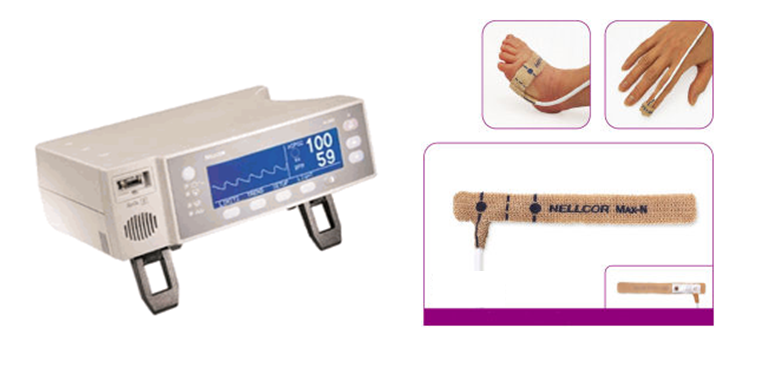
La saturation en oxygène ou satO2 : le dépistage systématique des cardiopathies chez le nouveau-né est mis en place dans les maternités avec la mesure de la saturation en oxygène aux membres supérieurs et inférieurs, dont le taux normal est > 97% avec une différence de moins de 3% entre les membres supérieurs et inférieurs. En cas d’anomalie de cette mesure, on réalise une échocardiographie pour rechercher une éventuelle anomalie cardiaque avant la sortie de maternité.
Saturomètre et capteur de saturation (satO2 )
Imagerie des cardiopathies congénitales
IRM
L’imagerie par résonance magnétique est d’un apport majeur pour l’expertise des cardiopathies congénitales, en particulier et surtout à l’âge adulte, quand la performance de l’échocardiographie est moins bonne.
L’IRM apporte des renseignements complémentaires indispensables à l’expertise, au suivi, au diagnostic et à la prise en charge et aux décisions thérapeutiques.
Elle permet :
L’évaluation anatomique des cavités et des vaisseaux
L’évaluation fonctionnelle : de la fonction myocardique
L’analyse des flux et des débits
La recherche de fibrose myocardique
La recherche d’ischémie par le test à l’adénosine
Sa réalisation est limitée chez l’enfant en raison de la nécessité de sédation.
Les indications principales concernent les ventricules droits systémiques (double discordance, transposition des gros vaisseaux avec switch atrial), les ventricules droits avec surcharge volumique (tétralogie de Fallot), les ventricules uniques, valvulopathies pulmonaires, aortiques, tricuspides, mitrales, les pathologies aortiques.
SCANNER
L’angio-scanner est une imagerie complémentaire permettant en particulier la visualisation des vaisseaux :
artères pulmonaires
veines pulmonaires
artères coronaires
aorte
fistules veino-veineuses et artério-veineuses
Sa réalisation est limitée par irradiation et injection de produit de contraste potentiellement néphrotoxique et allergisant.
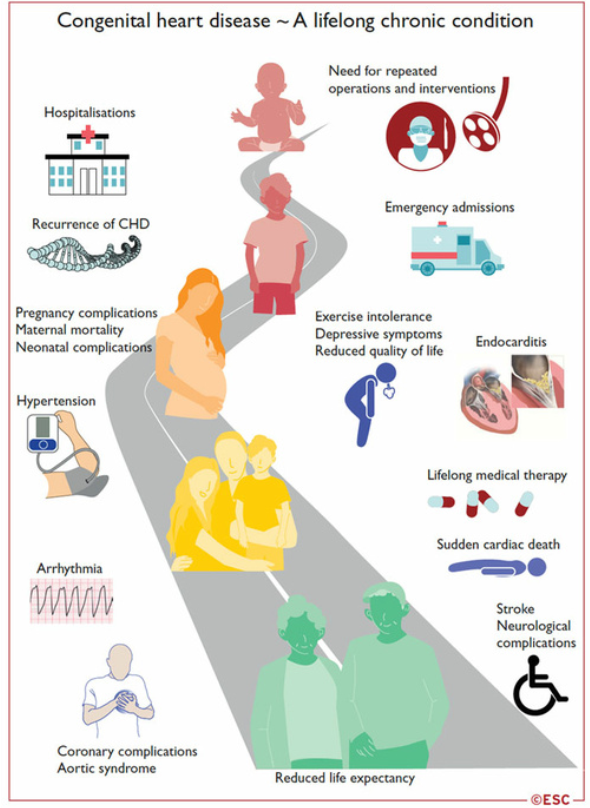
Cardiopathies congénitales à l’âge adulte
Plus de 90% des enfants avec une cardiopathie congénitale (CC) survivent à l’âge adulte
Le nombre d’adultes avec CC est maintenant plus important que le nombre d’enfants.
La plupart de ces patients ne peuvent pas être considérés comme guéris, et leur prise en charge est un processus qui dure toute la vie : de la réparation dans l’enfance, à la transition à l’âge adulte, au désir de grossesse, et à la prise en charge de complications tardives spécifiques à chaque CC.
Recommandations ESC 2020 : prise en charge des cardiopathies congénitales à l’âge adulte.
Transition à l’âge adulte
La phase de passage à l’âge adulte est importante et doit être anticipée et accompagnée, afin que le patient puisse recevoir toutes les informations sur sa cardiopathie, ses antécédents médicaux, ses traitements, son suivi et toutes les problématiques liées à sa cardiopathie.
Dans cet objectif, des programmes d’éducation thérapeutique ont été développés et mis en place dans les centres référents.
Au CCM : la transition se fait naturellement par une organisation basée sur une équipe médico-chirurgicale prenant en charge les mêmes patients du fœtus à l’âge adulte.
Complications des cardiopathies congénitales à l’âge adulte.
Dysfonction myocardique : ventricule gauche, ventricule unique, ventricule droit systémique
Coronaropathies
Insuffisance cardiaque
Arythmies
Endocardite infectieuse
Hypertension artérielle
Transmission et génétique des cardiopathies congénitales
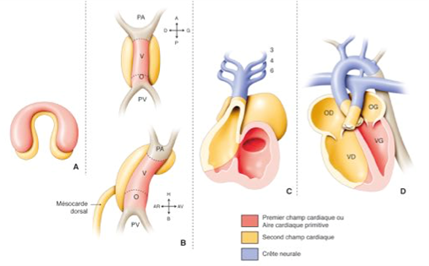
Embryologie du cœur normal : la formation du cœur a lieu pendant les 8 premières semaines de la vie fœtale, selon plusieurs étapes complexes successives partant du tube cardiaque primitif, avec des phénomènes de torsion et rotation et de cloisonnement :
Tube cardiaque primitif
Boucle : position des cavités oreillettes/ventricules
Formation des valves auriculoventriculaires
Cloisonnement des oreillettes
Cloisonnement des ventricules
Cloisonnement des vaisseaux
Les cardiopathies congénitales comportent un risque de transmission héréditaire par un mécanisme multifactoriel, à la fois facteurs génétiques et environnementaux. Dans la majorité des cas, soit 72%, aucune étiologie génétique n’est identifiable.
Facteurs génétiques et environnementaux : représentent 20% to 30% des cas.
Facteurs environnementaux : 2%
Maladie maternelle : diabète, rubéole, lupus érythémateux disséminé.
Consommation maternelle de traitements tels que : lithium, isotrétinoïne, anticonvulsivants, ou alcool et tabac.
Âge maternel : c’est un facteur à risque pour la trisomie 21, qui peut entrainer des malformations cardiaques.
Anomalies chromosomiques (aneuploïdies) : 10%
trisomie 21 ou syndrome de Down, trisomie 18, trisomie 13, monosomie X ou syndrome de Turner
Le risque de récidive des cardiopathies congénitales dans une famille dépend de la cause : négligeable dans les mutations de novo, de 2 à 5% dans les cardiopathies congénitales multifactorielles non syndromiques, et de 50% quand une mutation autosomique dominante est en cause.
Il est important d’identifier les facteurs génétiques pour évaluer le risque de récidive et pour guider le conseil génétique préconceptionnel.
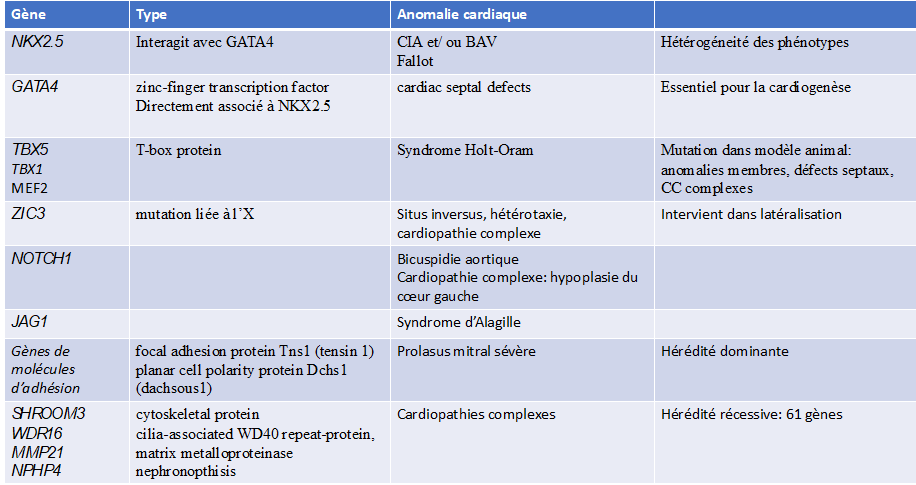
Exemples de mutations génétiques avec cardiopathies associées.
Contacter le +377 92 16 80 00 Les urgences relevant de la pathologie cardiovasculaire et thoracique sont acceptées sans restriction, 24 heures sur 24, 7 jours sur 7, dimanches et fériés compris.